There is still no cure for ALS, the most common form of motor neuron disease, or Lou Gehrig’s disease as it is commonly known in the USA. This is a devastating muscle wasting and paralysis disorder, usually fatal within a few years. With over 50 causative genes identified, why is a six-month extension of lifespan still the best we can offer patients?

Stephen Hawking (left) and Lou Gehrig both suffered from ALS (Image source: Wikipedia)
One important new line of thought is to approach ALS as a collection of rare disorders, not as a single disorder with one mechanism to block. Indeed, in a related rare disorder known as SMA (spinal muscular atrophy), direct targeting of its genetic mechanism has been extraordinarily successful.
But this approach is likely to be most effective with a mechanism that can be fully blocked and in patients where that mechanism is clearly active. Programmed axon death, studied in the Coleman lab, is a completely blockable mechanism. Removing SARM1, a protein that drives it, provides both (i) lifelong rescue in mice from a fatal genetic condition and (ii) complete rescue from a lethal neurotoxin. Both cause rare but fatal human conditions too. But does SARM1 have anything to do with ALS?

Our new preprint and another recent report make a compelling case that it does. What made this possible was three decades of research to understand the programmed axon death mechanism, the whole genome sequencing of thousands of ALS patients and controls by Project MinE, and the Ice Bucket Challenge and many other funders who supported Project MinE.

Image source: European Association for International Education
We knew enough about SARM1 to predict exactly whereabouts within it to look for the type of mutation most likely to cause disease – a gain-of-function. Many mutations altering residues in its inhibitory domain were exclusively present in patients in Project MinE, not in controls. Then, using enzyme assays for SARM1’s NAD degrading activity, we showed some of these mutations massively increase the activity of SARM1. We also tested the effect of one variant construct in neurons and found that it makes them extremely vulnerable. Another research group tested a different variant in mice with similar results.
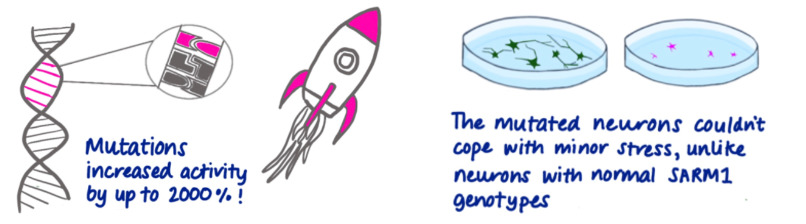
We then found more of these mutations, and a new one, in patients from other whole genome sequencing projects at AnswerALS, GENESIS and Genomics England and some in other motor nerve disorders, but still none in controls. The likelihood of this occurring by chance is negligible, with p<0.0000014.
And in an unexpected twist with potential therapeutic relevance, the hyperactivation of SARM1 in our assay was so high that it is hard to understand why it does not cause disease and death in very early life, even at or before birth. In contrast, most SARM1 variant patients developed disease only in middle- or old age and one as late as 71 years old. More studies are needed to solve this puzzle.
Other important questions remain too:
- These cases were sporadic, suggesting SARM1 interacts with other genes or environmental risk factors to cause disease, so what are these?
- Only around 0.2% of ALS patients have SARM1 coding gain-of-function variants so does SARM1 have wider involvement in ALS as GWAS studies and involvement of its likely upstream regulator STMN2 found by the Cleveland and Eggan groups suggest?
- Are ALS and these related disorders the only SARM1-associated diseases or is there much wider involvement as animal studies suggest?
- And most importantly for affected patients and their families, how can SARM1 blocking therapies be developed, can programmed axon death be blocked in the clinic in other ways, and how do we identify specific patients most likely to benefit from a successful drug blocking this mechanism for clinical trials and eventual treatments?
Research into programmed axon death to block SARM1 and other pathway steps has become an area of intense interest, but there is clearly much more to do.