How both genes and environment activate programmed axon death
It’s the question we ask about any disease: ‘nature or nurture?’. The answer is usually both, but in rare cases it’s one or the other. Progress in programmed axon death is beginning to show how SARM1 is activated by genes or environment, or by both acting together.
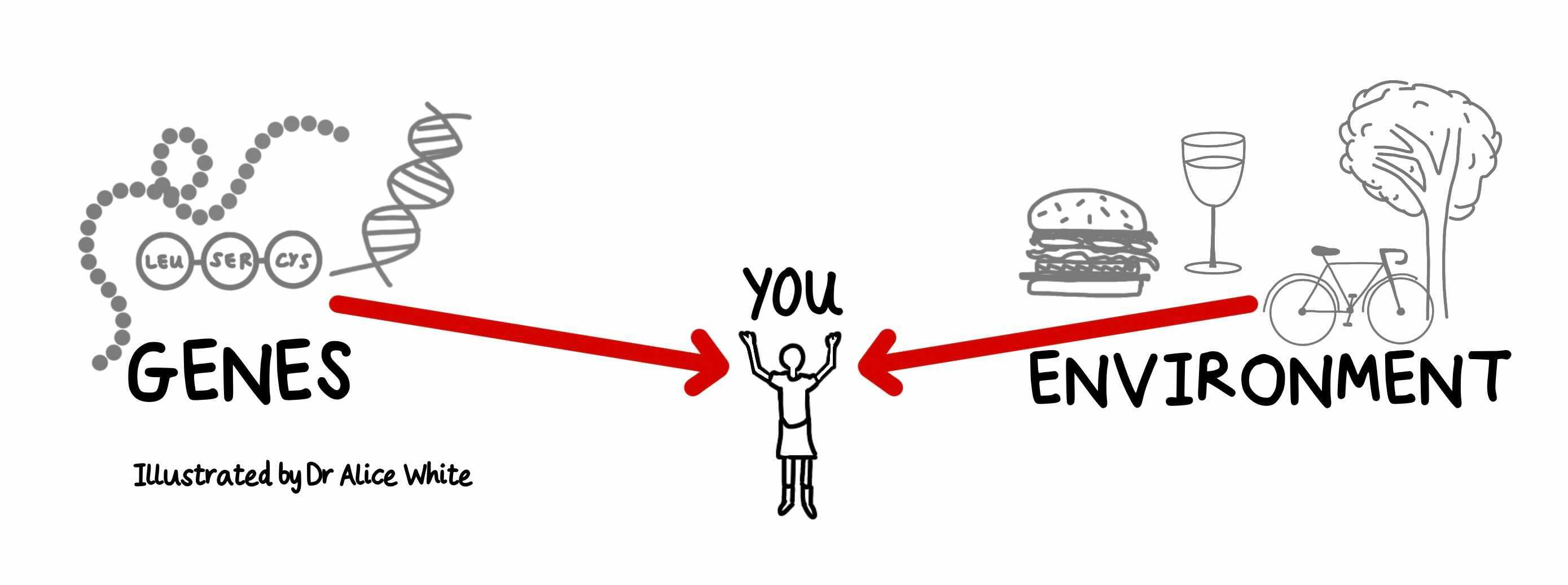
Genes, environment and programmed axon death
SARM1 is the central executioner of programmed axon death. It is an NAD degrading enzyme that kills axons. Its other enzyme activities may also contribute to neurodegeneration.
All of SARM1’s different enzyme activities are activated by the NAD precursor NMN. In 2015 and 2017, we showed that NMN promotes axon degeneration. Leading groups in China, Australia and USA then showed first that NMN directly activates SARM1 and subsequently that SARM1 senses the NMN:NAD ratio through competitive binding of these metabolites.
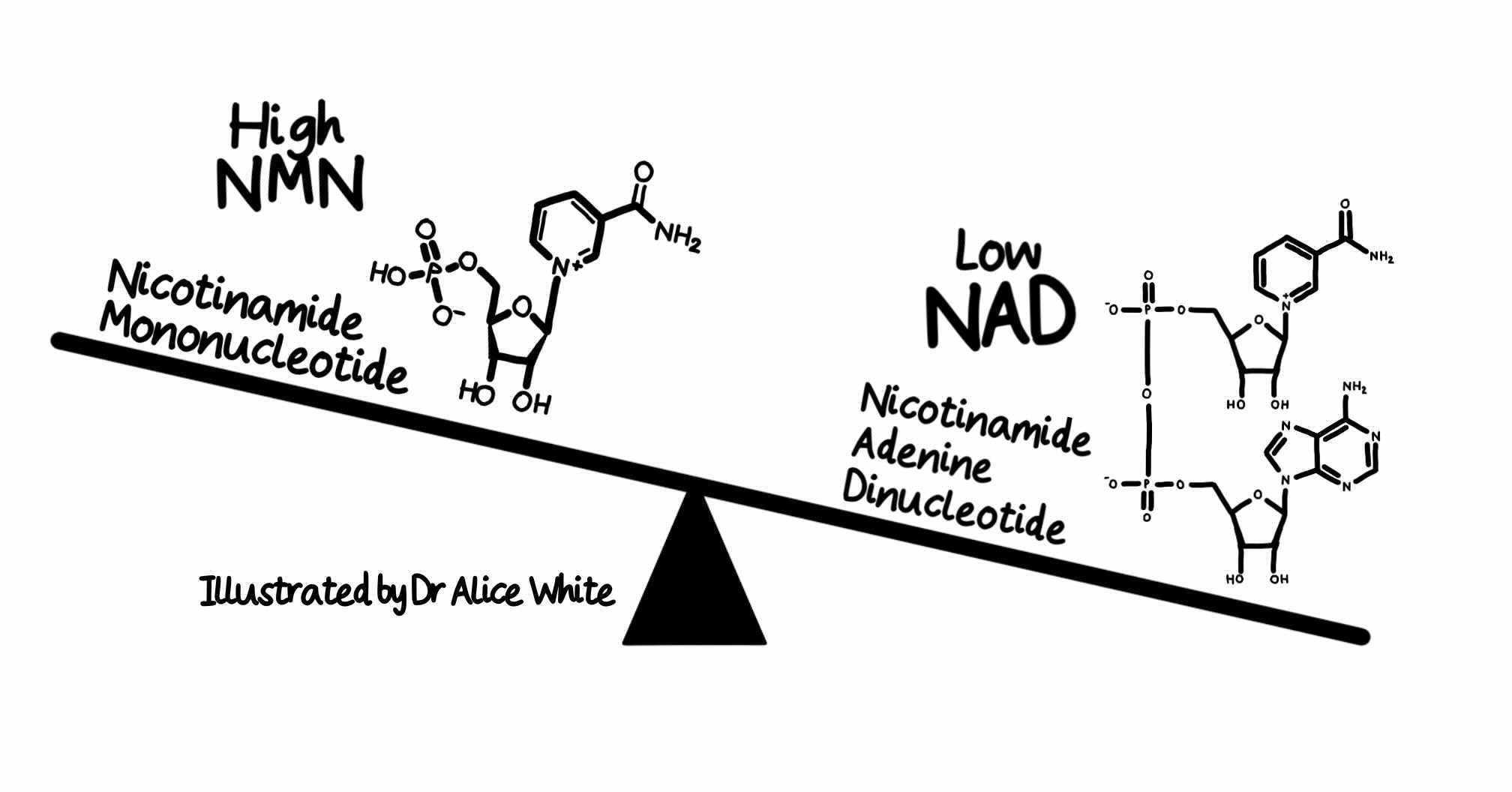
An elevated NMN:NAD ratio favours SARM1 activation. A preprint from our collaborators in Italy reports that this holds at physiological levels of both metabolites, and that there are additional endogenous regulators.
Five ways to activate SARM1
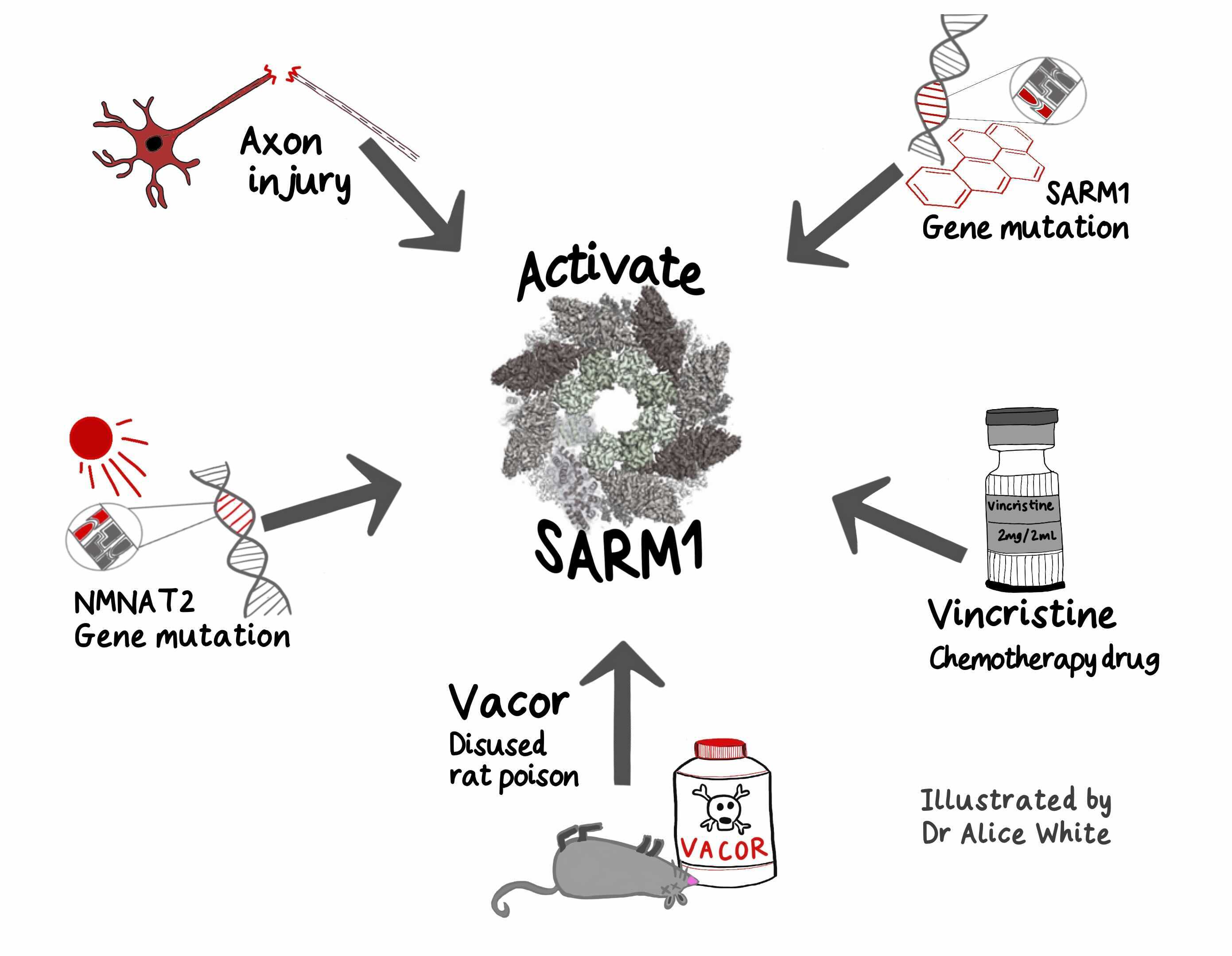
This deep understanding of basic mechanisms now reveals how both genes and environment impact this pathway to influence axon health. We know of at least five ways to activate SARM1. Some are genetic, some environmental and all are linked to human disease.
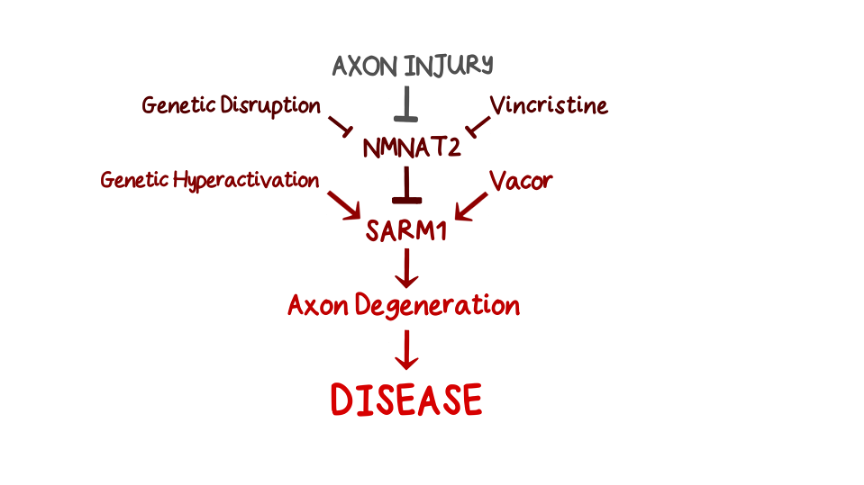
The NMN:NAD ratio rises when NMNAT2, an upstream regulator of SARM1 essential for axon survival, is lost or inactivated in axons. NMNAT2 is an enzyme converting NMN to NAD, keeping SARM1 inactive, but its levels fall dramatically after axon injury. So the first way to activate SARM1 is injury to nerves, spinal cord or brain.
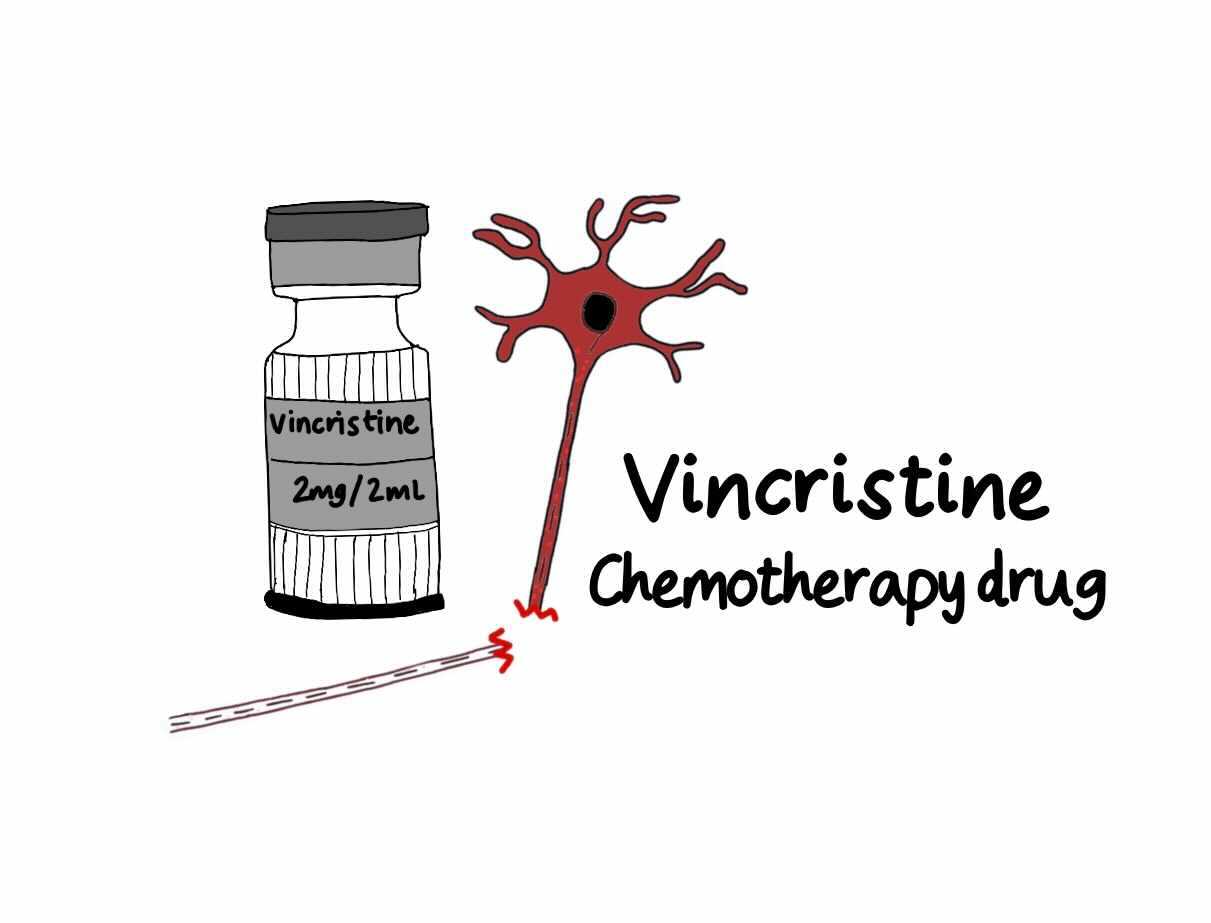
The second way is to disrupt axonal transport, impairing NMNAT2 delivery especially to the far ends of axons, for example in hands and feet. This happens for example with the cancer chemotherapeutic drug vincristine, which breaks microtubules. Animal studies indicate this causes SARM1-dependent peripheral neuropathy.
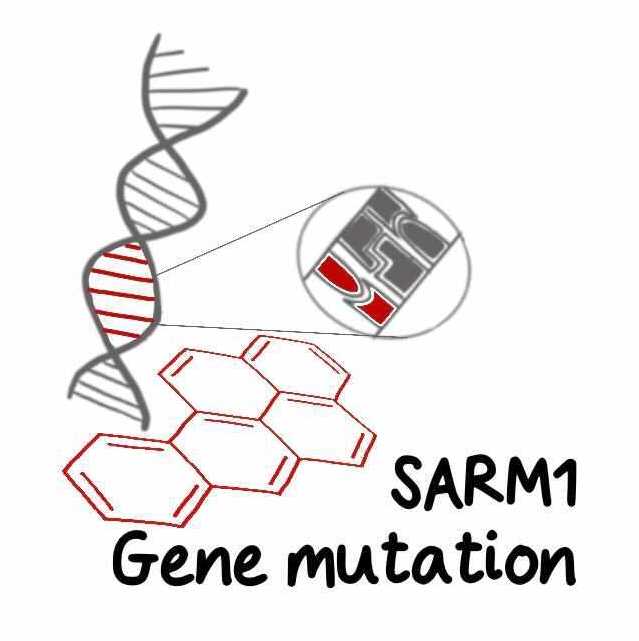
The third is genetic mutation of NMNAT2. In both humans [1,2] and mice this causes varying degrees of polyneuropathy involving sensory and motor axons and long axons in the CNS.

The fourth way is toxic activation of SARM1 itself. A preprint from our group reports SARM1 activation in toxicity by the disused rodenticide vacor. Vacor has a downstream metabolite that resembles NMN and very strongly activates SARM1. Vacor is associated with neuropathy in humans that is likely to be driven by SARM1 activation. Can other chemicals in our environment also activate SARM1? Another preprint subsequently reported similar findings with a second pyridine. Could low levels of such toxins activate SARM1 enough to kill a small percentage of our axons each year, with a cumulative outcome of age-related neurodegenerative disease? Would we know if this was happening?

And finally, gene mutations can massively hyperactivate SARM1. Preprints from ourselves and others report SARM1 hyperactivating mutations enriched in ALS patients and other motor disorders. Fascinating questions remain including whether other mechanisms activate SARM1 in ALS and how people survive at all with such highly active SARM1. Understanding this could help identify therapies.
Convergence
It is already clear that genes and environment can converge to activate programmed axon death. Low NMNAT2 expression makes axons more sensitive to vincristine. We now need to learn which other genetic or environmental risks combine with SARM1 mutation to cause ALS, or whether people with NMNAT2 or SARM1 gene variants respond differently to environmental risks such as some chemotherapeutics or nerve injury. These types of questions, moving beyond single causes, provide a way to understand, model and prevent sporadic disease.
And five ways to block SARM1
Encouragingly, there is rapid progress also in blocking SARM1 – again in five distinct ways. Its enzyme activity can be inhibited or it can be knocked down with antisense oligonucleotides. Elevating NAD using precursors may also help prevent SARM1 activation [1,2]. It is important to not also elevate NMN if NMNAT2 is low, but there are preliminary clinical successes potentially acting through this pathway.
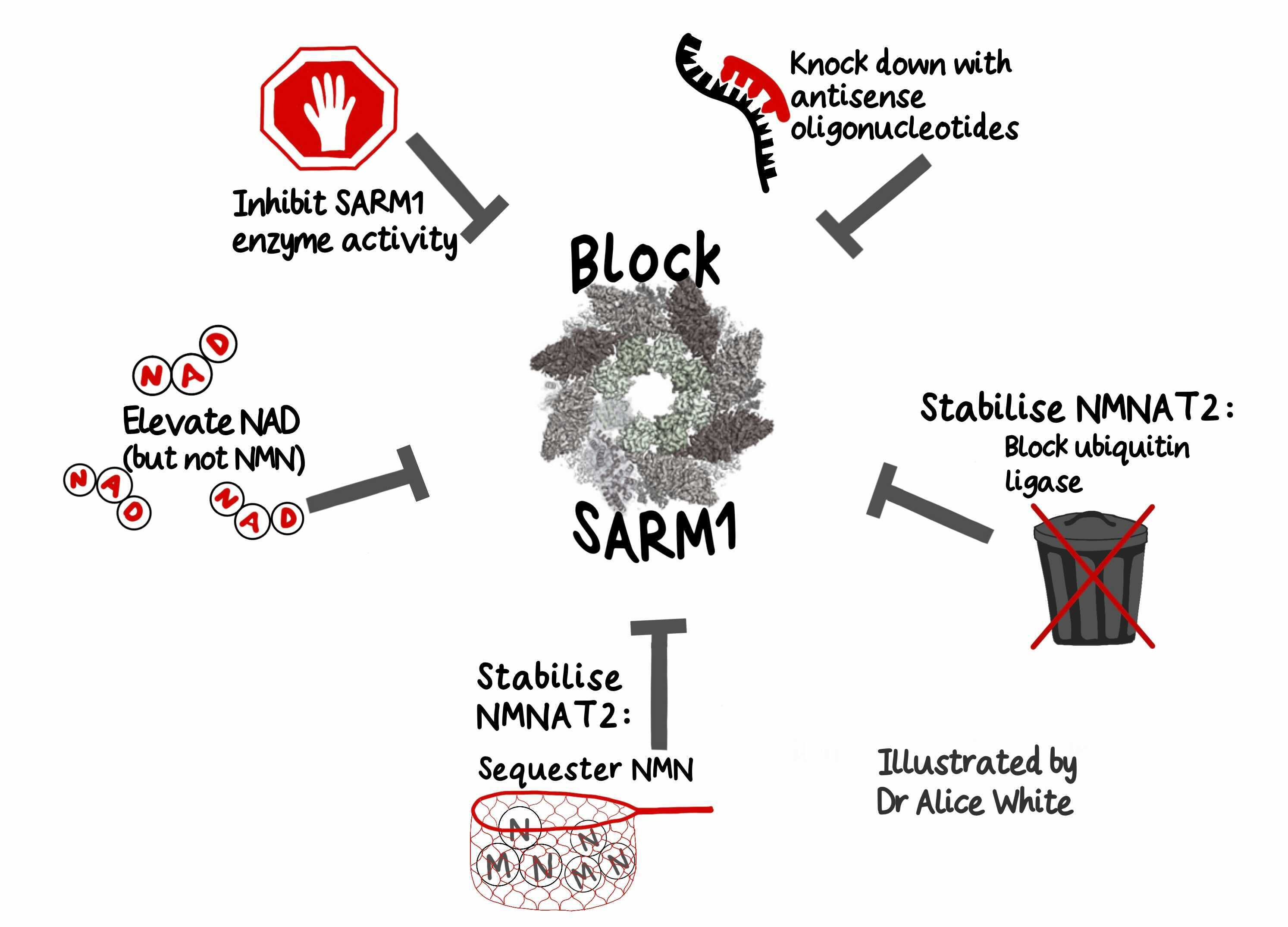
Two other approaches are very effective in animal studies but still at earlier stages of translation. One is to stabilise NMNAT2, for example by blocking its ubiquitin ligase and the other is to sequester NMN in another way.
Which one of these approaches, or which combination will be most effective clinically is an important future question, along with the vital step of matching the right drug to the right disease and right patients with personalized medicine. This field, which has come so far in 30 years, has developed an impressive momentum that promises years of exciting research ahead.